快速准确地预测分子特性,对于推进从材料科学到制药等领域的科学发现和应用具有重要意义。由于探索潜在选择的实验和模拟既耗时又昂贵,科学家们开始使用机器学习 (ML) 方法来帮助计算化学研究。但是,大多数 ML 模型只能利用已知或标记的数据。这使得准确预测新化合物的性质几乎是不可能的。
虽然标记的分子数据数量有限,但可行但未标记的数据数量正在迅速增长。
卡内基梅隆大学的研究人员思考他们是否可以利用这大量的未标记分子来建立ML模型,这种模型在属性预测方面比其他模型表现更好。他们最终开发了一个名为 MolCLR(Molecular Contrastive Learning of Representations with GNN) 的自我监督学习框架。MolCLR 通过利用大约 1000 万个未标记的分子数据,显著提高了 ML 模型的性能。
该研究结果以「Molecular contrastive learning of representations via graph neural networks」为题,于 2022 年 3 月 3 日发表在《Nature Machine Intelligence》上。
分子表征在新型化合物的设计中是基础和必不可少的。由于可能的稳定化合物的数量巨大,开发一种信息表示以概括整个化学空间可能具有挑战性。传统的分子表示,例如扩展连接指纹(ECFP),已成为计算化学中的标准工具。
近年来,随着机器学习方法的发展,数据驱动的分子表示学习及其应用,包括化学性质预测、化学建模和分子设计,越来越受到关注。
然而,学习这样的表示可能很困难。首先,分子信息很难完整地表示出来。其次,化学空间的大小是巨大的,这使任何分子表示都很难在可能的化合物中泛化。第三,分子学习任务的标记数据昂贵且远远不够。因此,大多数分子学习基准中的标签数量远远不够。在如此有限的数据上训练的机器学习模型很容易过度拟合,并且在与训练集不同的分子上表现不佳。
受益于可用分子数据的增长,自我监督/预训练的分子表示学习也得到了研究。
在这里,研究人员提出了 MolCLR(通过图神经网络进行表征的分子对比学习)来解决上述所有挑战。这是一种利用大量未标记数据(约 1000 万个独特分子)的自我监督学习框架。
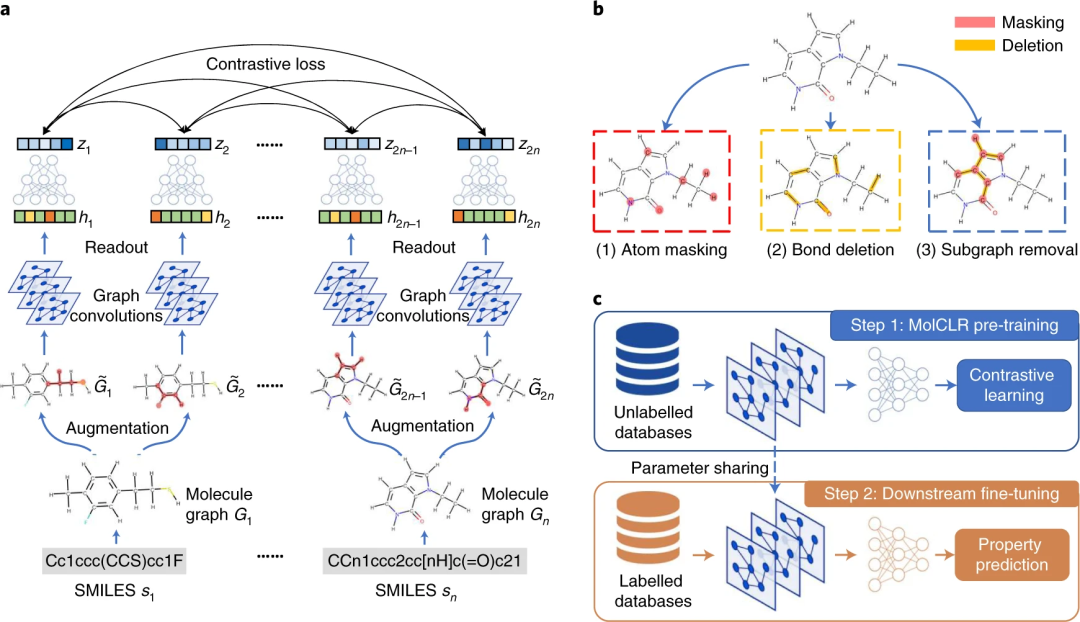
MolCLR 框架
MolCLR 模型是在对比学习框架的基础上开发的。来自正增强分子图对的潜在表示与来自负对的表示形成对比。整个管道由四个部分组成:数据处理和增强、基于 GNN 的特征提取器、非线性投影头和归一化温度标度交叉熵(NT-Xent)对比损失。
图示:MolCLR 概述。(来源:论文)
MolCLR 预训练的 GNN 模型针对分子特性预测进行了微调。与预训练模型类似,预测模型由 GNN 主干和 MLP 头组成,其中前者与预训练的特征提取器共享相同的模型,后者将特征映射到预测的分子属性中。微调模型中的 GNN 主干网络通过预训练模型的参数共享进行初始化,而 MLP 头则随机初始化。然后在目标分子特性数据库上以监督学习的方式训练整个微调模型。
在 MolCLR 预训练中,构建分子图并开发图神经网络编码器来学习可微表征。提出了三种分子图增强策略:原子掩蔽、键删除和子图删除。
在原子掩蔽中,消除了有关分子的一条信息。在键删除中,原子之间的化学键被擦除。两种增强的组合导致子图删除。通过这三种类型的变化,MolCLR 被迫学习内在信息并进行关联。
为了证明 MolCLR 的有效性,研究人员对来自 MoleculeNet 的多个具有挑战性的分类和回归任务的性能进行了基准测试。
表 1:不同模型在七种分类基准上的测试性能。(来源:论文)
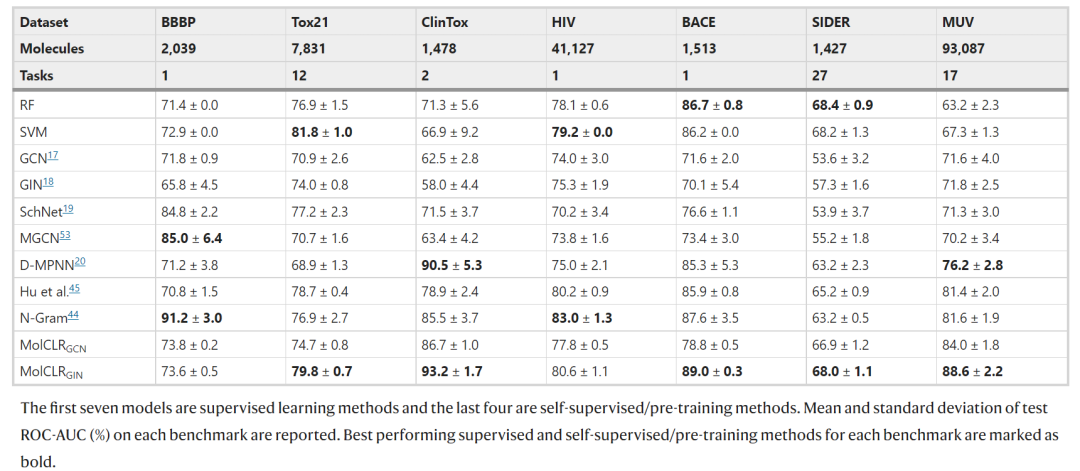
研究得出:(1)与其他自监督学习或预训练策略相比,MolCLR 框架在 7 个基准测试中有 5 个实现了最佳性能,平均提高了 4.0%。这种改进说明 MolCLR 是一种强大的自我监督学习策略,它很容易实现,并且对特定领域的复杂性要求很少。(2)与表现最好的监督学习基线相比,MolCLR 也表现出可匹敌的性能。在一些基准测试(例如,ClinTox、BACE、MUV)中,MolCLR 甚至超过了 SOTA 监督学习方法。(3)值得注意的是,MolCLR 在分子数量有限的数据集上表现非常出色,例如 ClinTox、BACE 和 SIDER。该性能验证了 MolCLR 学习了可以在不同数据集之间传输的信息表示。
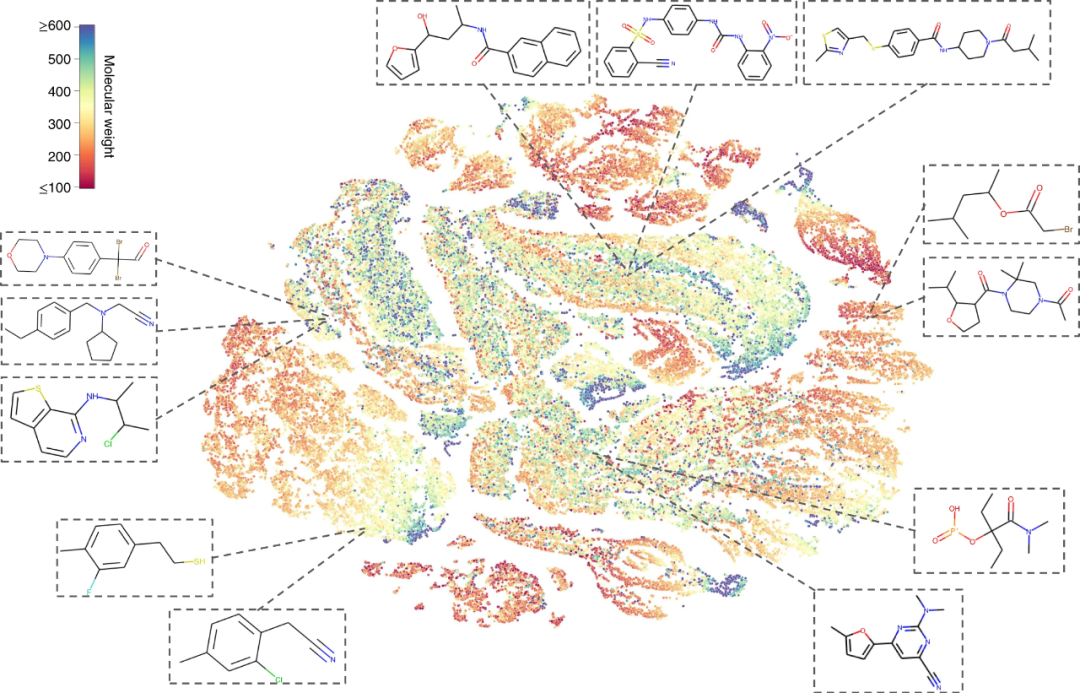
研究人员使用 t-SNE 嵌入检查由预训练的 MolCLR 学习的表示。t-SNE 算法将紧密的分子表示映射到 2D 中的相邻点。
MolCLR 学习了具有相似拓扑结构和官能团的分子的紧密表示。例如,顶部显示的三个分子具有与芳基连接的羰基。左下角显示的两个分子具有相似的结构,其中一个卤素原子(氟或氯)与苯相连。这说明即使没有标签,该模型也会学习分子之间的内在联系,因为具有相似特性的分子具有相似的特征。
图示:MolCLR 通过 t-SNE 学习的分子表征的可视化。(来源:论文)
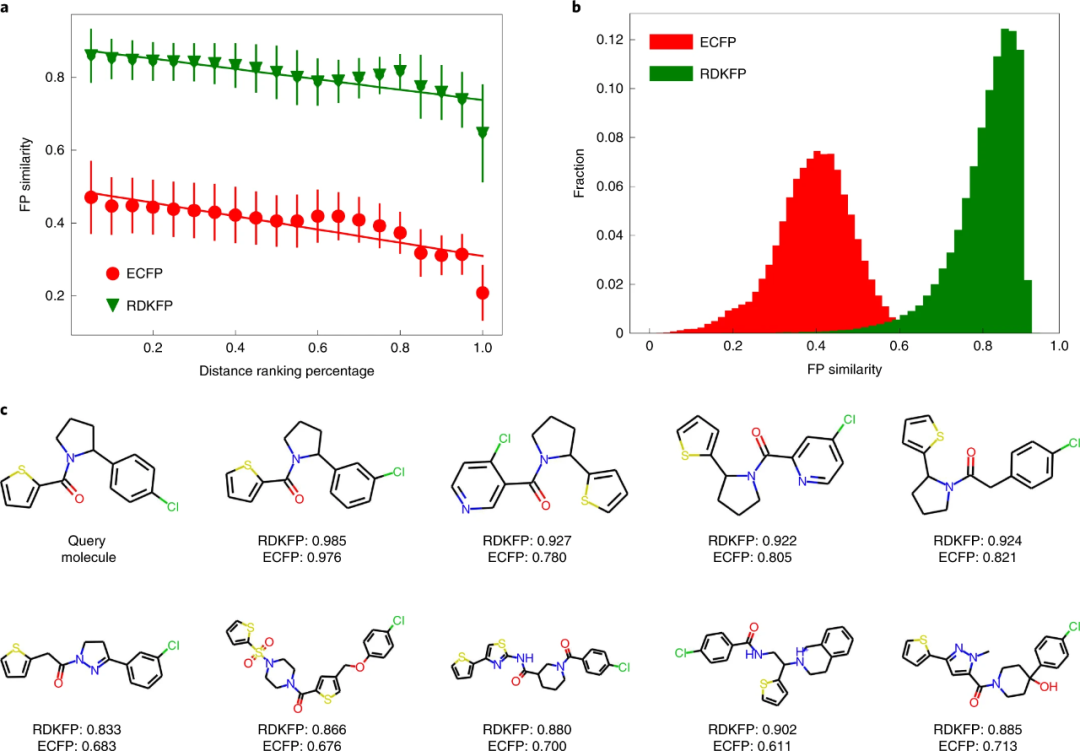
为了进一步评估 MolCLR,研究人员将 MolCLR 学习的表示与传统的分子 FP(例如 ECFP5 和 RDKFP)进行了比较。
图示:使用查询分子 (PubChem ID 42953211) 比较 MolCLR 学习表示和传统 FP。(来源:论文)
研究表明:通过对大量未标记数据的对比学习,MolCLR 自动将分子嵌入到代表性特征中,并以化学上合理的方式区分化合物。
「我们已经证明 MolCLR 有望实现高效的分子设计,」通讯作者、机械工程助理教授 Amir Barati Farimani 说。「它可以应用于多种应用,包括药物发现、能源储存和环境保护。」
作为未来的工作,有许多值得研究的方向。例如,GNN 主干的改进(例如,基于 Transformer 的 GNN 架构)可以帮助提取更好的分子表示。此外,自我监督学习表示的可视化和解释也很有趣。这样的研究可以帮助研究人员更好地了解化合物,有利于药物发现。
论文链接:https://www.nature.com/articles/s42256-022-00447-x
参考内容:https://techxplore.com/news/2022-03-machine-smarter-drug-discovery.html
人工智能 × [ 生物 神经科学 数学 物理 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。



文章评论